[screenshot from main page]

PriFi
works with a multiple alignment in the ClustalW format (suffix
.aln). You can either
[screenshot from main page]
By default, PriFi runs in the so-called intron mode in which at least one of the input sequences has to have specially annotated introns. This means that you have to manually edit one (or more) of your input sequences and insert X'es according to the table below, using a text editor of your own choice, before uploading the sequences. E.g., if you edit a genomic sequence with known intronic regions, you must cut out each intron and replace it with X'es. If you edit a coding sequence or an EST and you know the position and length of an intron, you must insert X'es at this position.
| XXX | intron, length <= 200 bp |
| XXXX |
intron, length 201 - 500 bp |
| XXXXX |
intron, length 501 - 1000 bp |
| XXXXXX |
intron, length > 1000 bp |
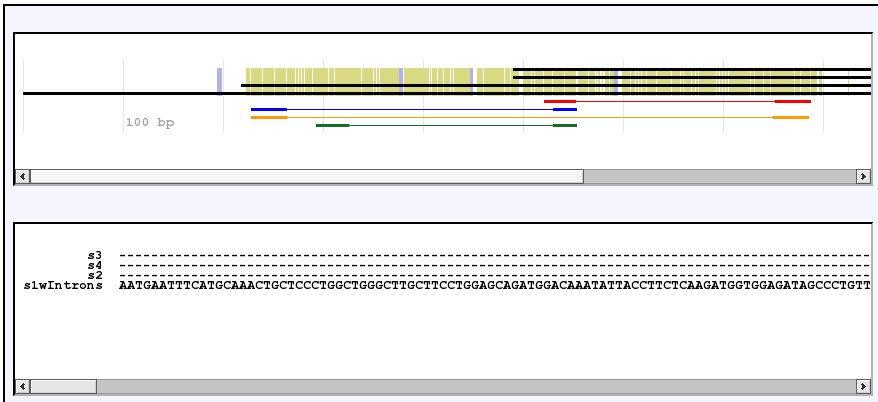
After a few seconds, PriFi returns a result. The result has three parts: two images and a textual report. The images show the alignment and the primers that PriFi suggests. Here is what they look like:
[screenshot from main page]
In the first image (the line-view), each sequence is represented symbolically by a line; in the second (the letter-view), the full sequences are written. In the main page you can scroll both images sideways. Input sequences are drawn in black while primer pairs are drawn each in its own color in both images. Perfect match alignment columns are highlighted in olive green, and in the line-view, introns are highlighted in grey.
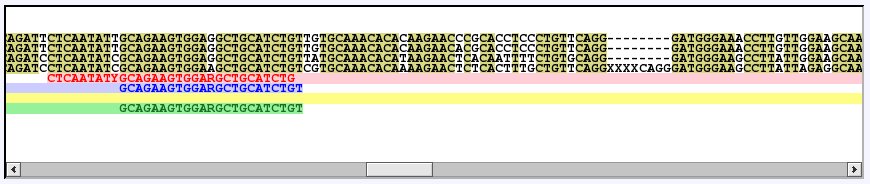
In the main page, scroll the letter-view until you find the start of the red forward primer:
[screenshot from main page]
Each primer pair is aligned with the input sequences so that you can see their annealing site. The expected resulting PCR product is indicated by a bar between the primers in a lighter shade of the same color. As you can see in the image, the red primer pair spans an intron - the intron is marked by XXXX in the bottom most input sequence (i.e. the fourth black sequence). When run in intron mode, PriFi only looks for primer pairs which span at least one intron.
PriFi also returns a textual report of the found primers. This report is shown beneath the alignment images, and it contains a detailed description of each primer pair. Here is the data for the red primer pair:
Note that the reverse primer is reverse complemented if you compare with its representation in the letter-view.
The report lists the properties of the primers; e.g. their lengths and melting temperatures, their distance to the closest primer, the expected PCR product length, and the number of A's and T's in the 3'-end tail. The report also lists the score of the primer pair as well as a specification of the score. E.g., the red primer pair is highly rewarded for its high melting temperatures and for "#sequences in primer alignments", which means that the primers are located in regions of the alignment where many sequences are represented. In fact, both primers are based on alignment regions where all four input sequences are represented, as indicated by the line "Avg. #sequences in primer alignments: 4.0 / 4.0". in the first part of the red report.
The only major penalty given to the red primer pair is for having four ambiguities in total. The total score is 160. Reports for the other primer pairs follow (see this section of the documentation for an explanation of how primer pairs are suggested).
The red primers yield a PCR product with an estimated length of 762 base pairs. Let's say that we're not interested in primers which yield PCR products shorter than 1000 base pairs. In order to change this parameter, press the "Configure" button on the PriFi main page. The configuration screen you'll get looks like this:
[screenshot from main page]
Each parameter name is a link which you can click to get a detailed explanation of the parameter's function (to try it, go to the main page). The "Minimum PCR product length" parameter is the seventh from the bottom, currently set to 450. On the main page, change it to 1000, then tick the demo alignment button above again and press the "Find primers.." button. Now PriFi finds a different set of primers; before, the best (red) pair had a score of 160 and was located sort of in the middle of the alignment. Now, the best pair is located at the left end of the alignment and has a score of 156:
[screenshots from main page]
The forward primer is now based on two sequences only, so the pair is less rewarded for the "#sequences in primer alignments". Nonetheless, PriFi believes this primer pair to be almost as good as the best one found with the previous default parameters. (In fact, if you look closely, you'll find that the red pair is identical to the blue pair of the first run, but since we changed the minimum PCR product length, it's now the overall best pair).
If you want to run PriFi again with the same parameter settings but on a different alignment, you have to modify the configuration again before uploading your data file.
Go to the main page, click the "Configure" button and set the "Minimum primer length" to 28 (fourth parameter from the top) and the "Maximum number of ambiguitity positions" to 0 (tenth parameter); then tick the demo alignment checkbox and press the "Find primers.." button. This time, you'll get
Thus, the chosen parameter settings are too strict and no primer pairs can be found which fulfill all criteria.
If you want to run PriFi in the general mode, you don't have to edit your input sequences and substitute introns with X'es. You press the "Configure" button and change the value of the last parameter, "Introns in sequences", to "no". Then you upload your data file and press the "Find primers.." button. In the general mode, primer pairs are not expected to span an intron (since now PriFi has no way of localizing introns), and all other criteria pertaining to introns are void, too.
Note that you have to actively change this parameter to run in general mode. If you forget to change it, you'll be running in intron mode while unaware of it, and PriFi will expect your sequences to contain X'es marking introns - and if none are found, PriFi won't find any primers for you (since in intron mode, primers have to span an intron). So make sure you set the "Introns in sequences" parameter to "no" before and each time you upload a data file with regular, unedited sequences.
Go to the PriFi main page