
(See also the tutorial.)
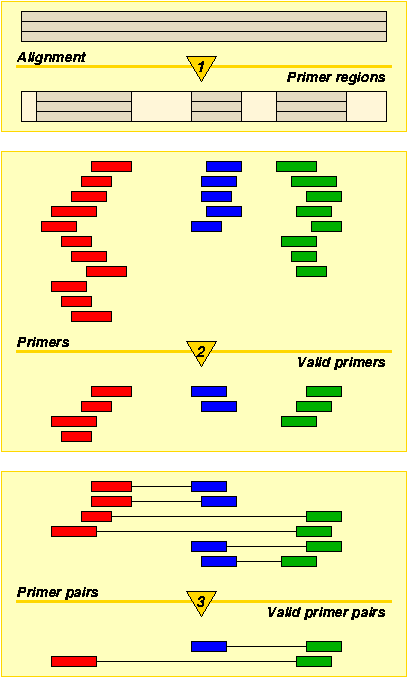
The aim of PriFi is to suggest a few primer pairs based on a DNA
sequence alignment, and to give an account of the quality of the
suggested primers.
PriFi lets the user either load a given alignment file (in the .aln
format), or, if the user has access to the alignment program Clustalw
(by the European Bioinformatics Institute), performs the
alignment from a multiple-sequence file (in the Fasta format). If not,
the user might obtain the alignment file using the web version of
Clustalw: http://www.ebi.ac.uk/clustalw/.
PriFi runs in one of two overall modes, either a general-purpose mode
or a so-called intron mode. In intron mode, the program expects one or
more of the sequences in the alignment to contain special intron
symbols: Before uploading his sequences, the user must substitute the introns of at least one of the
input sequences with X'es following this translation code:
| XXX | intron, length <= 200 bp |
| XXXX | intron, length 201 - 500 bp |
| XXXXX | intron, length 501 - 1000 bp |
| XXXXXX |
intron, length > 1000 bp |
(All parameters and criteria marked with * above become void when working in the
general mode rather than in intron mode).
A note on self-complementarity (quoting the PriFi paper):
Evaluation of self-complementarity is currently not supported. [..] PriFi is first and foremost an attempt to capture the, to some extent, intuitive yet successful practice of our laboratory for primer design, and here, self-complementarity is not taken into account.
Using the Oligo Calculator by Qing Cao, Warren, and Buehler (http://www.basic.nwu.edu/biotools/oligocalc.html), we found that around 10% of the primers had significant regions of self-complementarity that might in theory result in self-priming during PCR. However all these primers have worked well in the laboratory.
Further, one of PriFi's users (Anne Chenuil from Centre d'Oceanologie de Marseille) has sent me this comment:
Dear Jakob
I just read the results of your paper and find out that you actually
do not believe much in the self-complementarity criterion ....which I
find interesting as it confirms my personal experience: I used to
manually check thoroughly the primers and primer pairs for
complementarity and this gave me lots of complications, though I
observed that primers supposedly prone to these problems actually
worked well (as well as primers with a 3' mismatch nucleotide C, by
the way...)
[1] Jakob Fredslund, Lene H. Madsen, Birgit K. Hougaard, Anna Marie Nielsen, David Bertioli, Niels Sandal, Jens Stougaard, Leif Schauser
A general pipeline for the development of anchor markers for comparative genomics in plants
BMC Genomics 2006, 7:207
Go to the PriFi main page